近日,我院周先礼教授课题组在抗菌药物的设计、合成和成药性研究中取得新进展。相关研究结果以js33333金沙线路检测为第一单位,以“Design, Synthesis, and Biological Evaluation of Novel Arylomycins against Multidrug-Resistant Gram-Negative Bacteria”为题,发表在药物化学顶级期刊《Journal of Medicinal Chemistry》上,论文第一作者为周先礼教授指导的博士研究生张银勇,通讯作者为js33333金沙线路检测周先礼教授和上海药物所杨玉社研究员。

革兰氏阴性菌耐药危机日益严重,但相关新型抗生素的研发极其滞后,近50年来没有一款具有新靶点和新机制的抗耐药革兰氏阴性菌药物被批准上市。聚焦于全新靶点,研发结构新颖、作用机制独特且广谱强效的抗多药耐药革兰氏阴性菌药物是克服细菌耐药性的最有效方法。G0775作用于全新靶点细菌信号肽酶I(SPase I)(图1),因新结构、新机制和强药效,有望对抗细菌耐药性问题,目前正处在临床前研究。然而,G0775对临床分离多药耐药鲍曼不动杆菌和铜绿假单胞菌的活性较差,而且其在动物体内药代动力学性质不理想,限制了其应用。
针对以上情况,周先礼教授团队对G0775进行结构改造和优化,最终发现化合物138f(图1),对美罗培南耐药鲍曼不动杆菌和铜绿假单胞菌的MIC分别为0.125 μg/mL和4 μg/mL,比G0775增强8-16倍和4-8倍。在大鼠体内药代动力学研究中,静脉注射给药10 mg/kg,162(138f的游离碱,图1)的暴露量(AUC)和半衰期(T1/2)分别为141981 h·ng/mL和4.18 h,达到了G0775的2.3倍和2.8倍。在中性粒细胞减少的小鼠大腿感染模型中,感染菌株选择多药耐药铜绿假单胞菌,G0775不仅药效低下,剂量依赖性也缺失,而化合物162表现出显著优于G0775的体内药效,并且存在明显的剂量依赖性(图2)。分子对接结果显示(图3),162与LepB(大肠杆菌的SPase I)通过共价键结合,且空间取向和G0775基本相同,但具体结合模式有差异,这可能是162增强抗菌活性的原因。
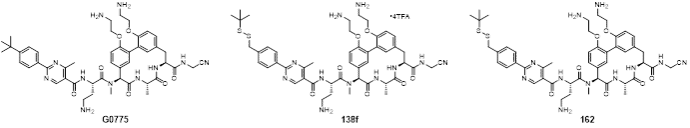
图1 G0775、138f和162的结构
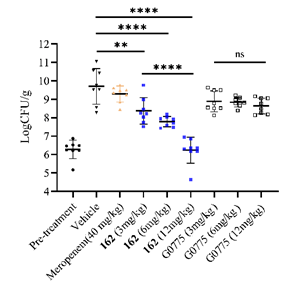
图2 美罗培南(meropenem)、162和G0775的小鼠体内药效
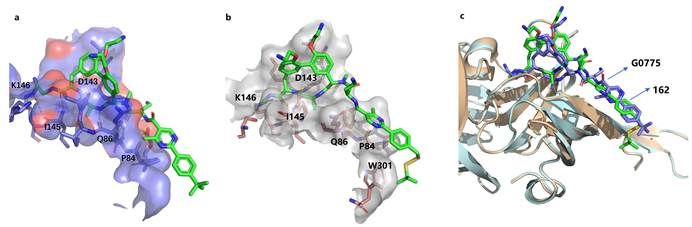
图3 (a)G0775和LepB(大肠杆菌的SPase I)复合物晶体结构(PDB:6B88);(b)通过分子对接预测的162与LepB的结合模式;(c)通过分子对接预测的162与LepB的结合模式与6B88的叠加构象
该工作发现了体内外抗菌活性和动物体内药代动力学性质均明显优于对照药G0775的候选化合物162,有望在对抗革兰氏阴性菌耐药性危机中发挥重要作用。
原文链接:https://doi.org/10.1021/acs.jmedchem.4c00018
文字/张银勇
{kind=link}
